
https://jameslyonsweiler.com/2017/11/28/biological-mechanisms-of-vaccine-injury-2-autoimmunity/ Nov, 2017 by James Lyons Weiler
THIS IS THE SECOND PART OF A SERIES OF ARTICLES ON THE BIOLOGICAL MECHANISMS OF VACCINE INJURY. The first of the series focused on the scientific evidence for (1) vaccine-induced mitopathy, (2) vaccine-induced persistent gliosis, (3) vaccine-induced endoplasmic reticulum failures (with both damage to detoxification pathways and to compromised immune systems via downregulation of ERAP1). Part 1: https://madisonarealymesupportgroup.com/2017/11/28/biological-mechanisms-of-vaccine-injury/
This article presents the scientific basis of autoimmunity from vaccines.
WHEN MOST OF THE KNOWLEDGE from basic science (cell culture and animal studies) seemingly falls apart in clinical trials, something is terribly wrong. Translational research falls apart when the basic science has it wrong, or when the clinical studies are flawed, fraudulent, or non-existent. Our governments have, in the past, relied heavily on the absence of evidence to assure the public on vaccine safety. “There are no studies” does not mean that the studies have been conducted… sometimes, it simply means “We don’t yet know”. They also rely on consistently over-cooked results that would raise the ire of any bona fide and ethical statistician.
Which is why it was most unscientific of the IOM to call for an end to science – after they ignored all of the basic studies on the question of a link between vaccines and autism – even though not all vaccines had been studied.
First, Immunology 101.
The mammalian adaptive immune system is a marvel of natural selection. Foreign proteins, or part of proteins (antigens) are taken up by antigen-presenting cells (APCs) including dendritic cells (DCs), where they processed into peptides and then loaded onto major histocompatibility complex (MHC) molecules for presentation to CD8 T cells via clonotypic T cell receptors (TCRs). (Stick with me, all of this is relevant, I assure you).
One type of T cell, the cytolytic T cells (Tc) can directly lyse a target pathogen cell. Tc’s are activated by MHC Class I on APCs. Another type of T cell, T helper cells (Th’s) are activated by MHC class II and release cytokines that have direct cellular effects themselves. Cytokines also activate macrophages, monocytes and B cells. B cells in particular express surface receptors that bind to surface antigens. When Th cells signal B cells, the B cells secrete antibodies that are supposed to be uniquely specific for the antigen responsible for the Th signal. Those antibodies can bind their specific targets alone, or they can also bind to and activate macrophages simultaneously, via the Fc receptor.
‘Priming’ of the immune system toward autoimmunity has been postulated. Vaccination, of course, often involves multiple exposures due to boosters. There are four mechanisms by which host infection by a pathogen can lead to autoimmunity. These are:
Molecular Mimicry. The pathogen carries elements that are similar enough in amino acid sequence or structure to self-antigen that the pathogen acts as a self-‘mimic’. In molecular mimicry, T or B cells activated in response to the pathogen also happen to be cross-reactive to self proteins. This can lead to direct autoimmune damage and false-positive “friendly-fire” activation of the immune system (via, for example, cytokine signalling due to the release of cytokines as result of autoimmune attacks on self proteins, cells, and tissues).
Epitope Spreading. A pathogen can also cause autoimmune disease via epitope spreading. In epitope spreading, damage to self-tissue occurs due either to the immune response to tissue infected by a persisting pathogen, or direct lysis of healthy cells by the pathogen. APCs take up antigens released from damaged tissue, initiating a self-specific immune response.
Bystander Activation. In this model, an indirect or non-specific activation of autoimmune cells is caused by the generally inflammatory environment that results from infection. The non-specific activation of one part of the immune system leads to the activation of other parts.
Cryptic Antigens. Foreign antigens can lead to autoimmunity via the activation of immunity to antigens that are not usually dominant – they are instead normally invisible to the immune system. It is generally described as an increase in “subdominant” antigens, and is usually attributed, like bystander activation, to the inflammatory environment that arises after infection. The involvement of cryptic antigens is considered likely when one observes increased protease production, and differential processing of released self-epitopes by APCs.
In the first article of this series, I reviewed the role of thimerosal as a specific inhibitor of ERAP1, a critically important protein for healthy immune systems. Read this description of the role of ERAP1 in APCs (Rock et al. 2010):
“ERAP1-deficient cells have reduced surface levels of MHC class I molecules and the peptide-MHC complexes that are made are less stable than on wild type cells… These results suggest that ERAP1 makes an important contribution both to the quantity and quality of peptides available for antigen presentation.”
Peptides presented by healthy cells come from normal autologous (self-generated) genes and are ignored because the immune system is tolerant to them. Aberrant peptide presentation due to low ERAP1 will lead to confusion of the immune system. When CD8 T-cells detect what appear to be foreign proteins because they are mis-processed, any tissue expressing the incorrectly trimmed peptides, either on their surface of during cell death is at risk of autoimmune attack.
The process of thimerosal-compromised ERAP1 failure will match the cryptic peptide model because peptides that do not normally initiate an immune response will appear to be highly immunogenic.
Anaphylaxis
When a total breakdown of normal controls against attacks against the self occurs, widespread autoimmune attacks lead to recurrent cycles of cytokine release and cell death – and any, or all of the processes described can be unleashed simultaneously. Organ damage, damage to blood vessels, and high fever can result in death.
A study of the rates of anaphylaxis reported to the Vaccine Safety Datalink (VSD) by McNeil et al. (2016) resulted in a finding of 33 confirmed cases of anaphylaxis from January 2009 through December 2011. Using the rate of 33 confirmed cases after 25,173,965 doses, they estimated the risk of anaphylaxis to be 1.31/million doses. It is interesting to note that 85% of the case of anaphylaxis had prior evidence of atopy (3 with prior anaphylaxis, 16 with asthma, and 9 with specific prior allergies).
The rate of 1.31 per million doses may seem small. However, a search of the VAERS (Vaccine Adverse Event Reporting System) database over the same time period (January 2009 through December 2011) reveals that 550 cases of anaphylaxis were reported to VAERS. VAERS is a passive reporting system, and users must acknowledge that the data cannot be used to attribute causality. Individuals who like to claim that VAERS is a sufficient and adequate system point to the positive finding of problems with a rotavirus vaccine. But unless causality can be established using data from VAERS, negative association results of no increase in risk of adverse events are suspect due to under-reporting.
Under-reporting of vaccine injuries is a serious issue for VAERS; estimates range from only 1% to 10% of adverse events captured. Using that range, the actual number of cases of anaphylaxis nationwide would be anywhere from 550 (20.1 per 1,000,000 doses), to 55,000 (2,000 per 1,000,000 doses). Although they are required to report all vaccine adverse events, there is no penalty to doctors who fail to report. Clearly, the post-market surveillance systems that are supposed to allow our scientists to detect upticks in vaccine injury (pharmacovigilence) do not work.
Guillan-Barré Syndrome (GBS)
The National Vaccine Compensation Program recently added GBS as a vaccine injury for which plaintiffs can be awarded compensation. GBS occurs when vaccines (or viruses) cause an autoimmune reaction against myelin proteins. These proteins act as insulation around nerves, and are essential for proper transmission of nerve impulses. GBS is not the only demyelination disease; in fact, most of the conditions that involve demyelination mostly differ in which tissue the syndrome represents (e.g., transverse myelitis (TM), multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS) and neuromyelitis optica (NMO).
Molecular mimicry was seen as a likely culprit way back in 1989:
“Thus, it is biologically plausible that injection of an inactivated virus, bacterium, or live attenuated virus might induce in the susceptible host an autoimmune response by deregulation of the immune response, by nonspecific activation of the T cells directed against myelin proteins, or by autoimmunity triggered by sequence similarities of proteins in the vaccine to host proteins such as those of myelin. The latter mechanism might evoke a response to a self-antigen, so-called molecular mimicry (Fujinami and Oldstone, 1989).”
Molecular mimicry due to exposure of individuals to proteins via infection by pathogens is non-controversial and widely accepted. Some examples of sequence-level similarity include the measles P3 protein and the human mylein basic protein (MBP), which shares at least 78% sequence identity:
MEASLES P3 …………………………….….EISDNLGQEGRASTSGTP….
HUM. MYLEIN BASIC PROTEIN ….EISFKLGQEGRDSRSGTP….
The likely effect of this level of sequence similarity is autoimmunity against the MBP, leading to change in the behavior of a host from mobile, and upright, to supine and sedentary – behaviors sure to elicit a social response by the host’s conspecifics, perhaps increasing the likelihood of transmission of the virus.
Some individuals are likely to more susceptible to autoimmunity due to exposure to proteins from pathogens owing to genetic variation that changes one or two amino acids in their self antigen sequence, making it more similar in sequence or in structure to the pathogen antigen.
The principles of autoimmunity from both viral infection and from vaccination have been absolutely demonstrated both in animals and in humans. McCoy et al. (2006) found that mice that were first exposed to a vaccinia virus encoding mylein protein, and then vaccinated against murine cytomegalovirus developed multiple sclerosis. In humans, cross-reactivity between MBP and a herpesvirus-6 protein has been reported (Tejada-Simon, et al. 2003). At the population level, a small increase in risk of MS and other demyelinating disorders due to vaccination (any vaccine) has been detected (Langer-Gould et al., 2014), but the authors concluded that the increased risk was real, but small.
A natural experiment in France occurred when the government brought Hepatitis B vaccination to the population. The uptake in HepB vaccine was followed by an increase in reported cases of MS to insurers (Figure 1). The government stopped purchasing the HepB vaccine, and as the remaining lots worked their way through the healthcare systems, HepB vaccine uptake declined, followed by a decline in the reported cases of MS (Figure 1). Other than a large randomized clinical trial that did not “correct for” or exclude individuals at risk of MS, this is the closest demonstration of causality between MS and vaccination that we can expect to see.
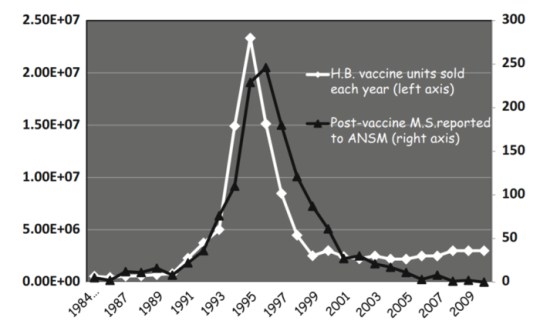 Figure 1. See Le Houézec, D. 2014. Evolution of multiple sclerosis in France since the beginning of hepatitis B vaccination. Immunol Res. 2014; 60(2-3): 219–225. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4266455/
Figure 1. See Le Houézec, D. 2014. Evolution of multiple sclerosis in France since the beginning of hepatitis B vaccination. Immunol Res. 2014; 60(2-3): 219–225. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4266455/
This outcome is not surprising, given the large amount of similarity shared by proteins in the HepB proteome and human neurological proteins (Ricco and Kanduc, 2010). Ample evidence exists in the form of cross-reactivity between antibodies against HepB myelin mimics and human myelin-related proteins (Bogdanos et al., 2005).
Individuals might experience varying degrees of severity of demyelination after vaccination due to genetic variation. Half of their myelin proteins and myelin oligodendrocyte glycoproteins might show low cross-reactivity because one of their parents had the canonical amino acid sequence, while the other half of their proteins might be more reactive due to genetic variation that alters the amino acid sequence in a way that increases the similarity to the pathogen’s matching peptide sequences. Further, over time, the progression and severity of demyelination could occur due to increased autoimmunity to the other version of the protein due to a process similar to bystander activation.
Aluminum Hydroxide and Autoimmunity
Vaccine risk denialists would have the public believe that the amount of aluminum in vaccines is not a threat to public health. With the exclusion of thimerosal in most pediatric vaccines came an expansion of the pediatric schedule, adding many vaccines that include aluminum hydroxide – with numerous doses.
It may surprise some, therefore, that aluminum hydroxide is routinely used in animal studies designed to model allergic rhinitis, (seasonal allergies), asthma (Elsakkar et al, 2016), food allergies (Ahren et al., 2014), autoimmunity, demyelination syndromes, and many other conditions (e.g., chronic prostatitis/chronic pelvic pain syndrome). Many of these studies use aluminum hydroxide to induce conditions identical to mysterious diseases in humans to show the efficacy of novel treatments.
Vaccine-Induced Asthma. Some of these studies take the additional step and use (all but in name) vaccine mimics – aluminum hydroxide combined with ovalbumin. Elsakkar et al. (2016), for example, used both aluminum hydroxide and ovalalbumin to induce asthma in mice to study the effect of adalimumab (aka Humira(R)). Other studies (e.g., Brandt et al., 2006) had previously demonstrated non-specific airway allergic reactions after dietary exposures to ovalbumin following exposure to aluminum hydroxide.
Vaccine-Induce Food Allergies. Up to 200 children die from anaphylaxis due to peanut allergies each year in the US, and the cost of caring for children with food allergies is estimated to be between $25 and $31 Billion – annually. In spite of this cost, no standard medical procedures exist advising parents against feeding their children peanuts or peanut butter after vaccinations.
In fact, the opposite approach is being tried. With limited scientific studies backing, the National Institutes of Health’s has, in 2017, recommended that parents feed infants as young as 4 to 6 months peanut puree to reduce the risk of the development of peanut allergies. This is based on one clinical study – with questionable results. While the study cites a marked reduction in the rate of evidence of peanut allergy in children with other allergies, only 82.9% of the SPT-positive group who were fed peanuts made it to the outcome determination, compared to 98% of the SPT positive arm who were not fed peanuts. The difference in patient attrition (98%-82.9%) in the SPT-positive group is a whopping 15% – much, much higher than the incidence of peanut allergy in the population (estimates range from 0.4%-1.4%). The drop-out rate difference would easily swamp the actual risk due to genotype.
Studies of treatments to reduce anaphylatic reaction (e.g., Shishehbor et al., 2010) use aluminum hydroxide. It known that that the simultaneous exposure of rats and mice to aluminum hydroxide and to foods such as egg. wheat, dairy, and nuts will likely cause the development of vaccine-induced food allergies. It would be remarkable if somehow humans did not also develop autoimmunity due to the combined effects of thimerosal ERAP1 suppression and the adjuvantive effects of aluminum hydroxide.
Rheumatoid Arthritis
A case report of three cases of RA after HepB vaccination (Gross et al., 1995) provides a clue that it is likely to be caused by vaccines in some people. Other similar reports exist, but population-level studies have failed to detect an association. Mouse models of arthritis exist that employ – you guessed it – injected aluminum hydroxide. So why would epidemiologic studies of samples of patients from the general population fail to detect association of RA and vaccines?
In humans, RA risk due to variation in the HLA genotypes should be a factor considered when designing studies of risk of RA due to vaccination. No study of the effect of vaccination on RA risk on individuals with HLA genotypes of known risk to RA has been conducted. These individuals are at highest risk. Like autism, RA risk has a genetic component, but certainly involves environmental factors. Karlson et al. (2013) found that the best model that could predict risk of RA includes both genes and environment. Unfortunately, vaccination status was not studied. If RA risk is enhanced via HLA genotype, only studies of that consider HLA RA risk (high and low) and vaccination status (vaccinated vs. unvaccinated) could answer the question of association whether HLA x vaccine interaction exists.
Molecular mimicry to pathogen proteins is suspected in RA (Singh and Karrar, 2014). Cross-reactivity is observed against mycobacteria protein and human cartilage (Holoshitz et al., 1986). Antibodies reactive to bacterial enolase have also been found in patients with RA (Lundberg et al., 2008). The specific human protein to which cross-reactivity was found is the human enolase protein (aka. CEP1, Enolase 1 (ENO1) alpha enolase 5-21). Rashid et al., (2007) found elevated antibodies that are cross-reactive to Proteus microbes – as well as to antigens that are not cross-reactive – in patients with RA.
Many patients with RA show cross-reactivity with citrullinated proteins. High among the likely candidates of self-antigen sources is the human protein vimentin. It is highly plausible that prior, or simultaneous exposure to vaccines and to non-pathogenic, or mildly pathogenic bacteria would be sufficient to induce (trigger) RA in genetically susceptible individuals. Studies focused on thorough searches for cross-reactive antibodies matching both human targets in tissues – and in pathways – are required.
It is worth noting that HLA genotypes confer risk of a diversity of autoimmune conditions, and thus studies of familial risk and environmental risk would be made more powerful by combining conditions influenced by similar HLA genotypes. It is plausible that the reason why whole population studies fail to detect association may well be that the association exists for an genetically identifiable minority of individuals.
It is also worth noting that some genetic variants in the ERAP1 gene, which is down-regulated by thimerosal, are associated with ankylosing spondylitis (AS) – an autoimmune form of arthritis in which the vertebrae of the spine can become fused. Individuals with RA are removed from studies of AS (e.g., Wang et al. 2012). Variants in ERAP1 are also associated with juvenile idiopathic arthritis (Hinks e al., 2011) and psoriasis (Strange et al., 2010).
Narcolepsy
One of the best examples of vaccine-induced autoimmunity is the sporadic occurrence of narcolepsy among families in Europe following H1N1 swine flu vaccination. Narcolepsy is a debilitating disorder in which sufferers suddenly fall asleep without warning. Researchers at Stanford University suspected the target protein was orexin (aka hypocretin), a protein secreted by cells in the brain that regulate the sleep/wake cycles. Their initial successful attempts to identify cross-reactive antibodies could not be reproduced, but then a study found strong cross-reactivity to influenza nucleoprotein and a receptor of hypocretin, hypocretin receptor 2 (Ahmed et al., 2015).
The vaccine that caused narcolepsy across Europe was Pandemrix, manufactured by GlaxoSmithKline. Other H1N1 flu vaccines with lower amounts of influenza nucleoprotein did not lead to narcolepsy. Incredibly, ministers in DPW in Britain fought to prevent a dozen children who developed narcolepsy due to vaccination with Pandemrix from being compensated for injury from the Vaccine Damage Payment Act. Fortunately for those individuals injured by Pandremix vaccination, the courts sided with the families.
Autism
Anti-brain protein antibodies are known to be found in children with autism (e.g., Zimmerman et al., 2007; Singer et al., 2008; Heuer et al., 2011). In 1988, a major report on pertussis and pertussis immunization (Cherry et al., 1988) reported:
“For more than 25 years, it has been known that pertussis vaccine is a reliable adjuvant for the production of experimental allergic encephalitis… This experimental allergic encephalomyelitis is mediated by sensitized lymphocytes rather than serum antibody mechanisms. Pertussis vaccine has also been used as an adjuvant in the following experimental autoimmune diseases: thyroiditis, myocarditis, glomerulonephritis, uveoretinitis, and hemolytic anemia.”
They then cited a lack of population-level association studies (i.e., no evidence) that pertussis vaccination results in these conditions in children. Gallup (2004) reminded the research community of this long-standing knowledge of the autoimmune-induced powers of DPT, citing his son’s case as an example. His son has “regressive autism and tested positive for myelin basic protein antibodies, has elevated measles antibody titers, T-cell abnormalities and colitis“.
Unless and until we study either “vaccine injured” vs. “not injured” to find biomarker signatures of moderate and serious adverse events, as was conducted with success for mild adverse events by Christian et al. (2015), or “likely susceptible” vs. “not likely susceptible” based on genetics, the study of the role of vaccines in inducing autoimmunity in humans will remain forever broken.
For now, the evidence that exists is simply overwhelming: vaccines can cause autoimmunity in some people, and we need (a) genetic screens to keep people out of harm’s way; (b) state bans on the use of unsafe peptides and epitopes in vaccines; (c) a test of Th1/Th2/Th17 balance prior to vaccination; (d) aluminum patch testing for aluminum sensitivity prior to vaccination. Consideration of HLA genotype for RA risk, or at least family history of autoimmune disorder, should be a matter of routine to reduce the likelihood of any vaccine-induced autoimmunity. The HLA-DR4 genotype is higher in frequency in autism (Lee et al., 2006).
We also need to start holding pediatricians accountable to treat all moderate and serious adverse events due to vaccination with appropriate follow-up testing, and emergent medical intervention. Failure to attend to their vaccine-injured patients should be reported as malpractice. The public does not need CDC, nor NIH, nor the AMA or AAP to acknowledge that vaccines induce autoimmunity in humans occurs to file complaints to the medical board or to their State Legislature.
NB: While completing this article, news of the publication of findings of record measurements of aluminum in the post-mortem autopsied brains of children with autism by Dr. Chris Exley’s team make a rather stunning bookend. The time has arrived for every person to let every member of the medical community know that aluminum is making us sick, and that one way or the other, they will be held responsible if they persist in their silence on the use of aluminum in vaccines. With all due respect, pediatricians, please join the Vaccine Risk Aware community in our calls for a reduction in the amount of aluminum in vaccines, a ban on thimerosal, and a ban on the use of unsafe peptides that match human proteins in vaccines.
References
Ahmed SS et al., 2015. Antibodies to influenza nucleoprotein cross-react with human hypocretin receptor 2. Sci Transl Med;7294):294ra105. doi: 10.1126/scitranslmed.aab2354.
Bogdanos et al., 2005. A study of molecular mimicry and immunological cross-reactivity between hepatitis B surface antigen and myelin mimics. Clinical and Developmental Immunology 12:217-224.
Cherry, J.D. 1988. Report of the task force on pertussis and pertussis immunization, Pediatrics 81:6 Part 11 (June 1988) Supplement pp 936-984.
Christian, LM et al. 2015. Proinflammatory cytokine responses correspond with subjective side effects after influenza virus vaccination. Vaccine. 33(29): 3360–3366.
Gallup, R. 2004. The new MMR? No! The Old DPT http://www.bmj.com/rapid-response/2011/10/30/re-no-old-dpt
Gross et al., 1995. Arthritis after hepatitis B vaccination. Report of three cases. Scand J Rheumatol. 24(1):50-2.
Heuer L et al., 2011. Association of a MET genetic variant with autism-associated maternal autoantibodies to fetal brain proteins and cytokine expression.Transl Psychiatry. 1:e48. doi: 10.1038/tp.2011.48.
Hinks A, Martin P, Flynn E, et al. 2011. Subtype specific genetic associations for juvenile idiopathic arthritis: ERAP1 with the enthesitis related arthritis subtype and IL23R with juvenile psoriatic arthritis. Arthritis Res Ther 13:R12.
Holoshitz J, 1986. T lymphocytes of rheumatoid arthritis patients show augmented reactivity to a fraction of mycobacteria cross-reactive with cartilage. Lancet. 2(8502):305-9.
Karlson, E et al., 2013. Association of Environmental and Genetic Factors and Gene-Environment Interactions with Risk of Developing Rheumatoid Arthritis. Arthritis Care Res (Hoboken). 65(7): 1147–1156.
Langer-Gould A 2014. Vaccines and the risk of multiple sclerosis and other central nervous system demyelinating diseases. JAMA Neurol. 71(12):1506-13. doi: 10.1001/jamaneurol.2014.2633.
Lee et al., 2006. HLA-DR4 in families with autism. Pediatr Neurol. 35(5):303-7.
Lundberg, K et al., 2008. Antibodies to Citrullinated a-Enolase Peptide 1 Are Specific for Rheumatoid Arthritis and Cross-React With Bacterial Enolase. Arthritis & Rheumatism 58:3009–3019
McCoy L et al., 2006. Multiple sclerosis and virus induced immune responses: autoimmunity can be primed by molecular mimicry and augmented by bystander activation. Autoimmunity. 39(1):9-19.
Rashid et al, 2007. Rheumatoid arthritis patients have elevated antibodies to cross-reactive and non cross-reactive antigens from Proteus microbes. Clin Exp Rheumatol. 25(2):259-67.
Ricco, R and D. Kanduc. 2010. Hepatitis B virus and Homo sapiens proteome-wide analysis: A profusion of viral peptide overlaps in neuron-specific human proteins. Biologics. 4: 75–81.
Rock KL et al., 2010. Proteases in MHC class I presentation and cross-presentation. J Immunol. 184(1):9-15. doi: 10.4049/jimmunol.0903399.
Singer HS et al., 2008. Antibodies against fetal brain in sera of mothers with autistic children. J Neuroimmunol. 194:165–172.
Singh A, Karrar S. 2014. The role of intracellular organisms in the pathogenesis of inflammatory arthritis. Int J Inflam. 2014:158793. doi: 10.1155/2014/158793.
Strange A, Capon F, Spencer CC, et al. 2010. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet 2010;42:985–90.
Tejada-Simon MV et al., 2003. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol. 53(2):189-97.
Wang, C-M et al., 2012. ERAP1 genetic variations associated with HLA-B27 interaction and disease severity of syndesmophytes formation in Taiwanese ankylosing spondylitis Arthritis Research & Therapy 201214:R125
Zimmerman AW et al., 2007. Maternal antibrain antibodies in autism. Brain Behav Immun. 21(3):351-7. Epub 2006 Oct 6.
Dr. Lyons-Weiler is a research scientist and author of three books, the latest of which is “The Environmental and Genetic Causes of Autism”. He is available for speaking engagements and book signing events at your location. To contact, follow on twitter @lifebiomedguru, email ebolapromo[at]gmail.com, and connect via LinkedIn https://www.linkedin.com/in/jameslyonsweiler
